Ein Risiko für die Qualität der Zulassung von SARS-CoV-2-Impfstoffen?
Bevor Arzneimittel oder – momentan sehr im Fokus – Impfstoffe vermarktet werden dürfen, müssen sie die langwierige Prozedur der Zulassung passieren. Für den europäischen Markt ist dafür die European Medicines Agency (EMA) mit Sitz in Amsterdam zuständig. Aktuell steht diese Behörde wegen der SARS-CoV-2-Impfstoffe unter erheblichem politischen und zeitlichen Druck. Nicht unbegründet machen sich einige Patienten deswegen Sorgen, ob ein in Eile zugelassener Impfstoff eine Gefahr für die Gesundheit sein könnte. Mit einem Blick in die Herangehensweise der EMA erkennt man, dass die Behörde nicht die Sicherheit riskiert, um die Geschwindigkeit zu erhöhen.
Für die Zulassung von SARS-CoV-2 Impfstoffen ist es wegen der dramatischen pandemischen Situation wünschenswert, diesen langwierigen Prozess zu beschleunigen. Aber ist dies ohne Weiteres möglich? Die FDA (Food and Drugs Administration, die Zulassungsbehörde in den USA) und die EMA vertreten eine eindeutige Position: Eine Zulassung kann erst dann erfolgen, wenn ausreichend Daten von guter Qualität vorhanden sind. Die FDA stellte sich daher gegen den Wunsch Donald Trumps, einen Impfstoff noch vor der Präsidentschaftswahl am 3. November 2020 zuzulassen. Nichtsdestoweniger ist der EMA bewusst, dass eine verzögerte Zulassung in der Pandemie erhebliche gesundheitliche und wirtschaftliche Schäden verursacht.
Ende November 2020 gab es weltweit nur eine vorläufige Zulassung eines SARS-CoV-2-Impfstoffs („Sputnik V“) in Russland. Dies beurteilte die EMA kritisch, da die Daten viele Fragen aufwarfen und sogar der Verdacht von Fälschungen bestand.
Auch Ungarn wollte den diskutierten Impfstoff Sputnik V nutzen und erteilte dafür die Notfallzulassung auf nationaler Ebene. Dazu erteilte die EU am 30. November grünes Licht. Eine Notfallzulassung erfolgt aber ohne tiefgehende Überprüfung der Daten und beinhaltet daher einige Risiken. Deswegen – anders als bei einer (bedingten) Zulassung, die die Europäische Arzneimittel-Agentur anstrebt – haftet im Falle von Impfschäden der Staat und nicht der Hersteller. Für die Arzneimittelhersteller ist das also eine willkommene risikofreie Markterschließung, die eigentlich nur in extremen Situationen zu gewähren ist. Diese Zulassung ist zudem zeitlich begrenzt und erlischt ohne die Möglichkeit, sie in eine bedingte oder ordentliche Zulassung umzuwandeln.
Am 2. Dezember 2020 erteilte Großbritannien eine Notfallzulassung für den Impfstoff BNT162b2 von Biontech/Pfizer, am 30. Dezember folgte die Zulassung der Vakzine AZD1222 von AstraZeneca. Damit demonstrierte Großbritannien seine Unabhängigkeit von der EU.
Entgegen den Erwartungen konnte am 21. Dezember die EMA in der Rekordzeit von drei Wochen ab Zustellung des Antrages die bedingte Zulassung für den COVID-19-Impfstoff BNT162b2 von Biontech/Pfizer und am 6. Januar für den Impfstoff mRNA-1273 von Moderna die Zulassung durch die EU-Kommission erwirken. Wie konnte die europäische Behörde die Zulassung dermaßen beschleunigen?
Was die Europäische Arzneimittel-Agentur macht
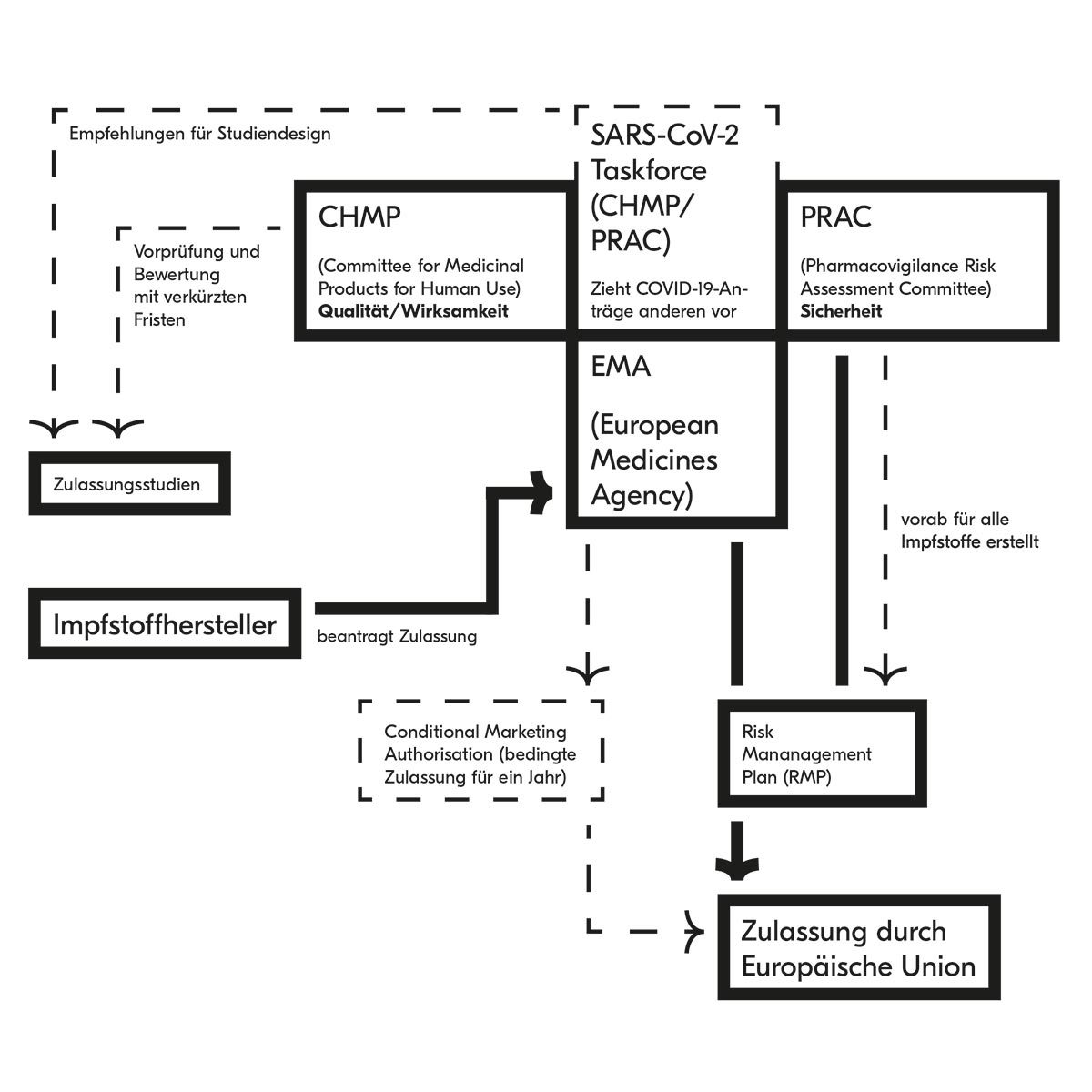
Aufgabe der EMA ist, die Qualität, Wirksamkeit und Verträglichkeit eines Arzneimittels zu prüfen. Bei der Zulassung attestiert sie einem Medikament eine positive Bilanz zwischen klinischen Vorteilen und Risiken – auf Basis der Evidenz, die sie von klinischen und nicht klinischen Studien gewinnen konnte. Dafür sind verschiedene Kommissionen zuständig, die aus Vertretern der nationalen Behörden, der Patienten, der Heilberufe und weiteren Experten zusammengesetzt sind:
CHMP
Committee for Medicinal Products for Human Use
PRAC
Pharmacovigilance Risk Assessment Committee
COMP
Committee for Orphan Medicinal Products, also für Krankheiten mit einer Prävalenz von weniger als 5 Patienten pro 10.000 Einwohner
CAT
Committee for Advanced Therapies, bzw. für Gen- und Zelltherapien, Tissue engineering
PDCO
Paedriatic Committee
Für einen Impfstoff sowie für klassische Arzneimittel sind zuerst der CHMP und der PRAC zuständig. Wenn die Arzneien auch für Kinder gedacht sind, wird auch der PDCO miteinbezogen. Der CHMP prüft zuerst die Wirksamkeit und die Qualität. Im Falle eines Impfstoffes muss die antragstellende Firma Studien vorweisen, die eine ausreichende Immunisierung über eine längere Zeit nachweisen. Gleichzeitig prüft der PRAC Daten zur Verträglichkeit und stellt einen Risk Management Plan auf.
Dieser Plan beinhaltet Elemente, die für die Sicherheit des Präparats relevant sind, und Maßnahmen, um die Sicherheit zu gewährleisten. Ist die Bilanz zwischen Wirksamkeit und Risiken positiv, erfolgt durch den CHMP eine Empfehlung an die Europäische Kommission, die die Zulassung für alle Länder der Europäischen Union erwirkt.
Der vom PRAC erstellte und wenn nötig adaptierte Risk Management Plan begleitet das Arzneimittel, solange dieses zugelassen ist. Der Hersteller ist verpflichtet, Sicherheitsdaten zu sammeln (z. B. unerwünschte Wirkungen) und in regelmäßigen Abständen dem PRAC zu berichten (Periodic Safety Update Report, PSUR). Diese Prozedur bedeutet für die Patienten, dass zugelassene Arzneimittel qualitativ einwandfrei, wirksam und sicher sind. Sie ist daher sehr wichtig, auch wenn dafür in der Regel mehrere Monate notwendig sind.
Die Gaspedale der EMA
Die EMA hat verschiedene Maßnahmen ergriffen, um eine Zulassung in kürzerer Zeit zu ermöglichen, ohne dass deren Qualität verloren geht. Zuerst ist bei der EMA eine Taskforce von Experten und Vertretern der Zulassungsbehörden gebildet worden, die in Sondersitzungen alle Themen rund um SARS-CoV-2-Impfstoffe behandelt. Die eingereichten Anträge werden anderen vorgezogen und ohne zeitliche Bindung bei den regulären Treffen der Kommissionen behandelt, welche üblicherweise monatlich erfolgen.
Diese Maßnahme kann schon per se eine Beschleunigung gegenüber dem Standard „accelerated assessment“ von maximal 150 Tagen bedeuten. Darüber hinaus sind die Fristen für die Evaluation der Experten verkürzt. Wenn normalerweise Experten bis zu 70 Tage für eine qualifizierte Antwort haben, so wurde diese Frist auf höchstens 20 Tage reduziert. Das bedeutet, dass die Experten SARS-CoV-2-Impfstoffe priorisiert bewerten müssen.
Die Taskforce hat auch Empfehlungen für die Ausführung von Studien herausgegeben, damit die Ergebnisse den Erwartungen der EMA bezüglich Wirksamkeit und Verträglichkeit entsprechen.
Eine weitere Maßnahme ist die Bereitschaft der Behörde, Daten aus abgeschlossenen Studien zu prüfen, noch bevor die Firmen alle Unterlagen für die Zulassung eingereicht haben. Normalerweise ist sehr viel Zeit für die Abarbeitung aller Daten notwendig. Durch eine Vorprüfung verkürzt sich die Zeit der Evaluation um Monate. Schon im November wurden Daten von Phase-III-Studien einiger Erfolg versprechender Impfstoffe durch Experten der EMA geprüft. Bei Einreichung des Antrags auf Zulassung können diese Daten dann ohne Zeitverzögerung in die finale Evaluation übergehen.
Des Weiteren hat der PRAC anhand der bekannten Risiken von Impfstoffen schon vorab den Risk Management Plan erstellt, der einheitlich für alle Anträge der Impfstoffkandidaten gelten wird. Auch diese Maßnahme reduziert die Zeit, die für die Prüfung der Zulassung notwendig ist. Insgesamt kann man also davon ausgehen, dass eine Zulassung von SARS-CoV-2 Impfstoffen nach Eingang von Anträgen sehr rasch erfolgen wird, ohne dass deren Qualität in irgendeiner Weise beeinträchtigt sein wird.
Als zusätzlichen finanziellen Ansporn verzichtet die EMA in einer pandemischen Lage auf die sonst sechsstelligen Zulassungsgebühren, sodass die Antragstellung wirtschaftlich erleichtert ist und auch für kleinere Firmen zu bewältigen sein wird.
Die Behörden wägen ab
Trotz dieser Maßnahmen bleiben einige Punkte zu berücksichtigen, die einer zu schnellen Zulassung von SARS-CoV-2-Impfstoffen entgegenstehen. Besonders RNA-basierte Impfstoffe, zu denen die Erfolg versprechendsten gehören, sind in ihrem Ansatz relativ neu. Nur wenige Produkte – z. B. gegen Lassa-Fieber oder Zika-Viren – wurden zuvor erfolgreich getestet. Um die Wirksamkeit und insbesondere die Dauer des Schutzes zu bestätigen, benötigt die Behörde klinische Studien der Phase III von ausreichender Dauer und mit einer ausreichenden Zahl an Patienten. Besonders wenn eine zweite Impfstoffdosis notwendig erscheint (wie bei den meisten COVID-19-Vakzinen), dauert die Studie entsprechend lang.
Der EMA bleibt allerdings auch die Möglichkeit einer bedingten Zulassung („Conditional Marketing Authorisation“). Diese kann sie erteilen, wenn die Notwendigkeit des Präparates für die allgemeine Gesundheit von erheblicher Bedeutung ist – was bei SARS-CoV-2 Impfstoffen der Fall sein dürfte –, die vorhandenen Daten keine besonderen Risiken erkennen lassen und zu erwarten ist, dass ausführlichere Daten in den darauffolgenden Monaten erhoben werden können.
In diesem Fall gilt die Zulassung für ein Jahr und kann entweder verlängert oder in eine endgültige Zulassung umgewandelt werden. Während dieser Zeit prüft die EMA in sehr kurzen Abständen (in der Regel alle drei Monate) die obligatorisch zu erhebenden Daten in der klinischen Anwendung auf Wirksamkeit und besonders auf Sicherheit. Zwar könnte ein solches Präparat möglicherweise riskanter als andere sein, doch die Behörden setzen dieses Risiko in Relation zu den Risiken, die resultieren, wenn kein Impfstoff Verfügbar wäre.
Fazit
Summa summarum stellt Zeitdruck für die Erarbeitung wissenschaftlicher Evidenz, die für die Zulassung notwendig ist, durchaus ein Problem dar. Aber durch entsprechende Maßnahmen können diese Risiken abgefangen werden – wie jetzt bei der EMA im Falle von SARS-CoV-2-Impfstoffen. Am Ende können wir davon ausgehen, dass bei einem von der EMA zugelassenen Arzneimittel die Risiken niemals höher sind als der Nutzen. Das ist im Falle einer Pandemie entscheidend.